What You Need To Know About Goldenhar Syndrome
The rare congenital condition known as Goldenhar syndrome is characterized by a variety of abnormal developments in association to the spine, ear and eye. Also known as OAV (Oculo-Auriculo-Verterbral Spectrum), this congenital condition was documented first by Maurice Goldenhar in 1952, a general practitioner and ophthalmologist. It is believed to affect 1 in every 3000 to 5000 births.
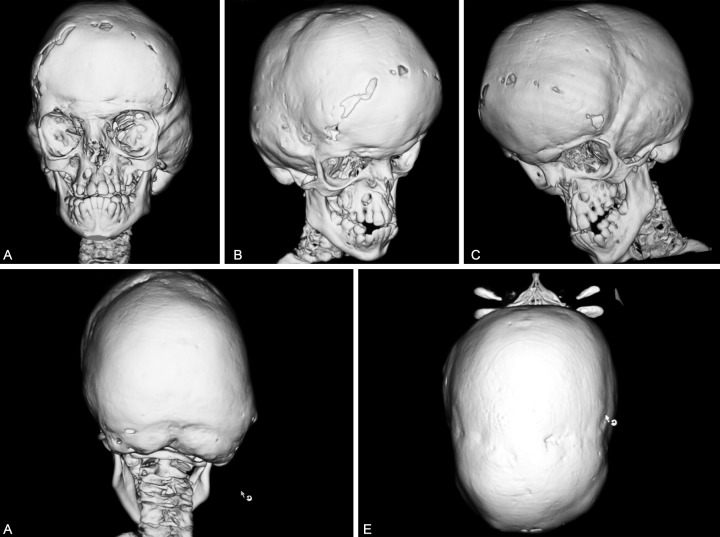
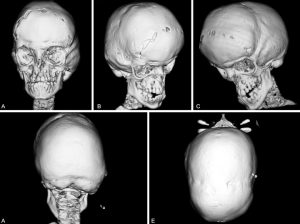
Babies that are born with Goldenhar syndrome will typically have totally absent or partially formed ears, spinal deformities like scoliosis and benign growths over the eyes. Goldenhar syndrome can also affect the child’s facial structure or organs such as the nervous system, lungs, kidney and heart. In the majority of cases, deformities will usually occur on 1 side of the child’s body.
One factor of Goldenhar syndrome has to do with hemifacial microsomia. This means the cheekbones and the jaw on 1 side of the child’s face will be undeveloped. The underdevelopment, combined with ear and eye anomalies, results in distinctive types of facial features for the children that have Goldenhar syndrome.
Rib cage and spine deformities are another common aspect of Goldenhar syndrome. In certain cases, the ribs or vertebrae inside the spine do not fully form, are fused abnormally or they are missing. About 50% of children that have Goldenhar syndrome will present a type of congenital-scoliosis. The spine anomalies result in pulmonary disorders and incomplete growth.
Symptoms And Signs
The symptoms associated with Goldenhar syndrome will vary, but can include the following features:
• Spinal abnormalities that lead to Kyphosis, scoliosis or both
• Cardiac defects
• Abnormal rib structure that includes fused or missing ribs, this leads to decreased lung function, thoracic insufficiency and poor growth
• Hearing loss, that typically occurs in 1 ear
• A variety of craniofacial disorders that include:
– Cleft palate or cleft lip
– Hemifacial microsomia, which involves tissues on both sides or one side of the child’s face that is underdeveloped, especially in areas that affect the jaw, mouth and ear areas
– A mouth that is wide, with one side that is slightly higher compared to the other
– Benign growths or cysts over or around the eyes known as ocular-dermoid cysts
– Totally absent or partially formed ears known as microtia
• Central-nervous-system defects
• Respiratory issues
• Urogenital and kidney issues
Treatment
Due to the fact that Goldenhar syndrome has the potential to affect a number of the body systems, the treatments for this condition will vary. In certain cases, monitoring with care is all that will be necessary. For other cases, surgery will be required in order to address certain aspects of this condition.
A variety of complications in association to Goldenhar syndrome will be evident when the baby is born and are treatable while the child is still young. Some of these examples include craniofacial anomalies, hand disorders and club feet.
Other types of complications of Goldenhar syndrome can only start to show up or become problematic as the child grows. These issues usually include joint disorders, dislocated hips and spinal deformities.
Each child’s condition will be different, which means a treatment plan will be determined in regards to a case-by-case basis. Treatments can include non-surgical options like physical therapy and bracing, or surgical treatments such as implanting and then expanding growing-rods to stabilize the child’s spine as they grow or spinal fusion.
Follow-Up Care
A child will Goldenhar syndrome must be monitored by orthopaedic physicians into their adult years. If the child has undergone spinal-fusion surgery, they will have appointments scheduled with an orthopaedic surgeon a week after the surgery and then 3 to 6 months post-surgery.